IMAGING INTEGRATO DI UN CASO DI SINDROME DI
McCUNE-ALBRIGHT
N. Zaccheo, A. Tirelli, N.C. Sforza, L. Falcone
Azienda U.S.L. BA/4
Servizio di Radiologia P. O. "Di Venere" - BARI
Direttore Angela Patrizia Garribba
INTRODUZIONEL'associazione di displasia fibrosa poliostotica (POFD), macule cutanee iperpigmentate ed endocrinopatie multiple con pubertà precoce, è stata dapprima descritta nel 1922 da Weil
[1] e, successivamente, meglio delineata nel 1937 da McCune e Brunch [2] e poco dopo da Albright [3]. Da allora la sindrome di McCune-Albright (MAS) è rimasta patologia affascinante e controversa, rappresentando, nella forma classica, una rara affezione, più frequente nel sesso femminile, con incidenza del 3% dei casi di POFD [3, 4, 5, 6].
Quest'ultima, di per sé, domina il quadro sindromico con alterazioni strutturali ossee sclerotiche, pagetoidi e pseudocistiche, singole o associate, prevalentemente o esclusivamente localizzate ad un emischeletro e condizionanti per sede, numero e stadio di sviluppo l'età di insorgenza e la variabilità della sintomatologia [3, 5, 6, 7, 8].
Riportiamo un caso di MAS pervenuto alla nostra osservazione in acuto per comparsa in regione fronto-orbitaria di una tumefazione dolente a rapido accrescimento di sospetta natura neoplastica sovrapposta e diagnosticato con TC, RT, RM sulla base delle alterazioni scheletriche caratteristiche. Ne riteniamo interessante la descrizione per la sporadicità della patologia e la peculiare modalità clinica di manifestazione.
DESCRIZIONE DEL CASORagazza di 14 anni; da alcuni giorni febbricola, cefalea intensa, esoftalmo, edema palpebrale, disturbi della visione e comparsa in regione fronto-orbitaria sinistra di tumefazione sottocutanea dura, dolente con iperemia della cute sovrastante.
Alle indagini emato-chimiche: lieve leucocitosi ed incremento della VES; GH, prolattinemia, cortisolemia, TSH, T3, T4, calcemia totale, fosforemia nella norma.
Nell'anamnesi remota: pubertà precoce a 8 anni.
All'esame obiettivo: normale corporatura, oftalmoplegia sinistra, macule cutanee brunastre a bordi irregolari e contorno definito in regione sternale.
La TC, eseguita prima e dopo m.d.c. in urgenza, documenta, in corrispondenza della tumefazione fronto-orbitaria, la presenza di formazione espansiva, osteolitica, similcistica, disomogenea per livello fluido-fluido, che assottiglia e rigonfia il tavolato cranico, estrinsecandosi in fossa cranica anteriore e verso il globo oculare, dislocandolo. L'indagine rileva inoltre, altre lesioni osteolitiche similtumorali cranio-facciali e diffusa sclerosi delle ossa della base cranica compatibili con displasia fibrosa (fig. 1A-B).
I radiogrammi dello scheletro in toto, eseguiti ai fini di stadiazione di malattia, rivelano un interessamento poliostotico, politopo, prevalente all'emisoma sinistro. Le localizzazioni craniche appaiono, in sede diploica, di aspetto litico benigno simil-cistico con rigonfiamento dei tavolati cranici ed associate ad osteosclerosi di vicinanza. Nelle ossa del massiccio facciale predomina l'aspetto osteosclerotico. A carico dei restanti segmenti scheletrici, con predilezione per il tratto meta-diafisario delle ossa tubulari, prevalgono aree rotondeggianti, a margini netti e talora delimitati da un sottile cercine osteosclerotico, di radiotrasparenza priva di strutturazioni interne, tuttavia in alcune sedi perifericamente e centralmente all'area radiotrasparente si delineano zone addensate simil-osteosclerotiche, sfumate, con sovvertimento strutturale della compatta e della spongiosa. Il tratto prossimale delle diafisi femorali è alquanto slargato ma non sono documentabili rime fratturative (fig. 2A-B-C).
La RM cranio-encefalica prima e dopo m.d.c., espletata ai fini di una pianificazione terapeutica, completa l'iter diagnostico meglio definendo i rapporti anatomici della localizzazione ossea cranica pseudocistica, verosimilmente complicata da fenomeni degenerativo-emorragici per livello fluido-fluido (fig. 3A-B).
Quest'ultima lesione è stata sottoposta ad intervento chirurgico con successivo controllo istologico.
Istologicamente la lesione era costituita da frammenti di osso spugnoso e compatto, nonché da aree di tessuto fibroso riccamente cellulare con frammiste trabecole osteoidi a prevalente distribuzione marginale. Nel contesto del tessuto fibroso si rilevava altresì la presenza di una discreta quota di cellule giganti plurinucleate e istiociti a citoplasma schiumoso, spesso con disposizione a vallo periluminale. Tali elementi deponevano per la diagnosi di displasia fibrosa in involuzione cistica con marcate modificazioni reattive secondarie.
CONSIDERAZIONI La MAS è una sporadica condizione morbosa genetica non ereditaria, ad eziologia sconosciuta, prevalente nel sesso femminile.
Una precoce mutazione post-zigotica del gene GNAS1 del cromosoma 20 (posizione q13.2) attiva con modello "a mosaico" la subunità alfa della proteina eterotrimerica G, accoppiata ai recettori di membrana. Tale proteina, modificata, stimola l'adenilciclasi a produrre cAMP, segnale mitogenico per alcuni gruppi di cellule. Le principali conseguenze sono una proliferazione cellulare ed una produzione ormonale abnorme [9,10].
Nei pazienti con POFD la sindrome si manifesta con gravità clinica variabile nel 3% dei casi. La POFD può associarsi, tuttavia, solo a manifestazioni cutanee o endocrine rispettivamente nel 20% e 5% dei casi [2, 3, 4, 7].
Macule cutanee iperpigmentate, a bordi irregolari e contorno definito, interessanti più o meno estese aree del corpo con predilezione di quelle inferiori, possono già essere presenti alla nascita o comparire nei primi anni di vita [7, 11].
La precoce maturazione sessuale può comunemente dipendere da attivazione del sistema ipotalamo ipofisario, come nel nostro caso (pubertà precoce), ma più raramente può essere espressione di una iperfunzione ovarica primitiva (pseudoubertà precoce) [9,12]. La comparsa di cicli mestruali nella femmina e di spermatogenesi nel maschio precede di solito le lesioni ossee variabilmente da 6 mesi a 9 anni (nel nostro caso 5 anni e 9 mesi); nel maschio la precoce maturazione sessuale si accompagna frequentemente ad acne e particolare odore [4,12]. In entrambi l'accrescimento scheletrico encondrale può essere anomalo; all'inizio accelerato, ma poi precocemente concluso con conseguente riduzione della statura [8]. In aggiunta al precoce sviluppo sessuale possono coesistere multiple endocrinopatie, in particolare: ipertiroidismo e/o struma, gigantismo, sindrome di Cushing, iperprolattinemia, rachitismo ipofosfatemico, adenomi pituitari [3, 4, 7].
La POFD è l'elemento portante della sindrome [2, 3, 4, 5, 7, 8]. Le ossa più frequentemente colpite sono il femore prossimale, le ossa mascellari, la tibia, le coste e l'ala iliaca ed al di fuori delle zone interessate, le ossa hanno aspetto normale. Nelle ossa lunghe la sede d'inizio è la metafisi o il tratto meta-diafisario, con possibile estensione a gran parte o alla totalità del segmento scheletrico, con eccezione delle epifisi. Ad un esame macroscopico estemporaneo, le ossa colpite appaiono espanse ma meno consistenti e pesanti che di norma; l'aumento dello spessore è per lo più esteso a tutta la lunghezza della diafisi e della metafisi e, talora, essendo più frequenti nelle forme monostotiche, possono aversi tumefazioni circoscritte meta-diafisarie, affusate, neoplastiformi (corrispondenti radiologicamente a zone di aspetto pseudocistico) che corrispondono a masse fibrose rivestite da corticale sottile e divise in concamerazioni da sepimenti ossei. Il quadro istologico è dominato dalla proliferazione fibrosa che comincia dalla cavità midollare e dagli spazi midollari e si estende, lungo i canali di Havers, alla corticale che viene erosa dall'interno verso l'esterno e trasformata in spongiosa. Nelle sedi delle lesioni si rileva un tessuto fibroso ricco di fibroblasti frammisto a caratteristiche sottili trebecole ossee od osteoidi distribuite in modo variabile e disordinato e talora a cellule giganti plurinucleate a tipo osteoclastico e cellule schiumose cariche di lipidi [7, 8].
I segni clinici della malattia sono variabili a seconda soprattutto della sede delle lesioni, del loro numero e del loro stadio di sviluppo: modico dolore, tumefazione, fratture patologiche anche ripetute (85% dei casi), deformazioni scheletriche specie degli arti inferiori. Nelle coste la tumefazione è particolarmente evidente e, se a rapido accrescimento e in mancanza di altre localizzazioni, può giustificare il sospetto di una neoplasia maligna. La trasformazione maligna delle lesioni ossee fibrose per lo più in osteosarcoma o ancor più raramente in condrosarcoma e fibrosarcoma è evento assai poco comune [5, 7, 8, 13]. Il caso clinico giunto alla nostra osservazione ha posto una serie di interrogativi, soprattutto da questo punto di vista. Gli esami diagnostici effettuati (TC, RM, RT), integrati da un'accurato raccordo anamnestico e dall'esame obiettivo della paziente, hanno comunque consentito il corretto inquadramento diagnostico.
TC e RM hanno, inoltre, permesso con un certo margine di sicurezza di escludere la degenerazione neoplastica.
A tal proposito preme sottolineare che le indagini radiologiche danno risultati diversi in base all'estensione delle lesioni e anche alla quantità e distribuzione dell'osso neoformato nell'ambito dei focolai patologici [8]. Gli aspetti radiologici della displasia fibrosa possono essere ricondotti a tre patterns distinti: pagetoide, sclerotico e simil-cistico. Il pattern pagetoide è il più comune (53% circa dei casi), riscontrandosi usualmente in pazienti di età superiore ai 30 anni, ed è caratterizzato da regioni ossee espanse con aree alternate di osteosclerosi ed osteorarefazione. I patterns sclerotico e pseudocistico, si riscontrano in pazienti più giovani e si ritiene siano precursori della forma pagetoide. Il primo è caratterizzato da regioni ossee espanse con aumentata densità a tipo "ground glass"; il secondo, da lesioni rotondeggianti ed ovali simil-cistiche con orletto sclerotico. Tali aspetti sono documentabili con il massimo dettaglio informativo con TC, specie a livello delle ossa temporali e del massiccio facciale [6,14].
Le alterazioni ossee della displasia fibrosa possono essere riconosciute anche con RM che consente una migliore caratterizzazione tessutale correlabile alla quantità dei componenti la struttura delle lesioni (trabecole ossee, cellularità, collagene, modificazioni cistiche ed emorragiche). L'osso appare ispessito e nelle sequenze T1W e T2W rispettivamente ipointenso (100%) e ipo (38%) o iperinteso (62%). Dopo somministrazione di gadolinio si può assistere (88%) ad un modesto enhancement dell'osso displasico, centralmente (73%) o con ring periferico (27%). Il pattern pseudocistico con fenomeni di degenerazione adiposa e di emorragia intralesionale si riscontra nel 15%-21% dei casi talora con problemi diagnostico differenziali con il tumore a cellule giganti, specie nei casi di lesione unica in ossa tubulari [6,14,15].
L'indagine RM, nel nostro caso, è risultata decisiva ai fini della pianificazione terapeutica per una resezione chirurgica il più conservativa e radicale possibile della lesione pseudocistica i cui rapporti anatomici sono risultati non ben valutabili con i soli piani di scansione assiali e con le ricostruzioni MPR fornite dalla TC, in relazione alla sua particolare sede. Lo studio RM, inoltre, per l'elevata risoluzione spaziale e di contrasto ha permesso anche un'accurata valutazione delle strutture contigue oculari e di fossa cranica anteriore escludendone il loro coinvolgimento.
CONCLUSIONI In conclusione riteniamo che l'imaging radiologico integrato, fondamentale nel riconoscimento della patologia, risulti oltremodo indispensabile nella valutazione delle forme complicate.
BIBLIOGRAFIA- Weil H: Pubertas praecox und Knockenbruchigkeit. Klin Wschr 1: 2114, 1922.
- McCune DJ, Brunch H: Osteodystrophia fibrosa; report of a case in which the condition was combined with precocious puberty, pathologic pigmentation of the skin and hyperthyroidism, with review of the literature. Am J Dis Child 54: 806-848, 1937.
- Albright F, Butler AM, Hampton AO et al: Syndrome characterized by osteitis fibroma disseminata, areas of pigmentation and endocrine dysfunction with precocious puberty in females; report of five cases. N Engl J Med 216: 727-746, 1937.
- Benedict PH: Endocrine features in Albright's sindrome (Fibrous dysplasia of bone). Metabolism 11: 30-45, 1962.
- Nager GT, Kennedy DW, Kopstein E: Fibrous displasia: a review of disease and its manifestation in the temporal bone. Ann Otol Rhinol Laryngol 91(suppl 92): 5-52, 1982.
- Lambert PR, Brackmann DE: Fibrous dysplasia of the temporal bone: the use of computerized tomografy. Otolaryngol Head Neck Surg 92: 461-467, 1984.
- Lanza G.: Ossa. In: Lanza G (ed) Anatomia patologica sistematica, pag 2075-2077, Edizioni Piccin Nuova Libraria, Padova, 1985.
- Fiore-Donati L: Patologia ossea. In: Calì A., Fiore-Donati L (eds) Anatomia patologica generale ed applicata, pag 1087, USES-Edizioni Scientifiche, Firenze, 1988.
- Weinstein LS, Shenker A: G protein in human disease. Clin Biochem 26 (5): 333-338,1993.
- Lania A, Mantovani G, Spada A: G protein mutation in endocrine diseases. Eur J Endocrinol 145 (5): 543-559, 2001.
- Davies JH, Barton JS, Gregory JW, Mills C: Infantile Mc-Cune-Albright syndrome. Pediatr Dermatol 18 (6): 504-506, 2001.
- Rieth KG, Comite F, Shawker TH, Cutler GB: Pituitary and ovarian abnormalities demonstrated by CT and ultrasound in children with features of the Mc-Cune-Albright syndrome. Radiology 153: 389-393, 1984.
- Kaushik S, Smoker WR, Frable WJ: Malignant trasformation of fibrous dysplasia into chondroblastic osteosarcoma. Skeletal Radiol 31(2): 103-106, 2002.
- Lupescu I, Hermier M, Georgescu SA, Froment JC: Helical CT and diagnostic evaluation of cranio-facial fibrous dysplasia. J Radiol 82 (2): 145-149, 2001.
- Fibrous dysplasia: MR imaging charateristics with radiopathologic correlation. AJR 167(6): 1523-1527, 1996.
ICONOGRAFIA
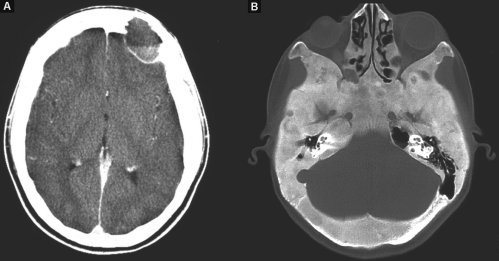 Fig.1
Fig.1 TC dopo iniezione di m.d.c.:
A) presenza in sede fronto-orbitaria sinistra di lesione espansiva, osteolitica, similcistica, disomogenea per livello fluido-fluido che assottiglia e rigonfia il tavolato cranico, estrinsecandosi in fossa cranica anteriore;
B) la finestra dell'osso rivela una diffusa sclerosi delle ossa della base cranica ed altre aree osteolitiche simil-tumorali di minori dimensioni.
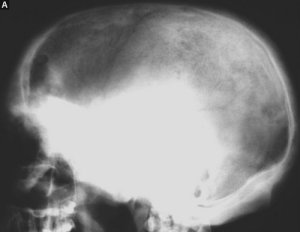
 Fig.2
Fig.2 RT:
A) diffusa sclerosi della base cranica con iniziali aspetti pagetoidi della teca ed aree litiche frontali;
B) rigonfiamento bolloso dell'arco di torsione della IV e dell'arco posteriore della VIII costa di sinistra;
C) multiple aree iperdiafane confluenti di aspetto cistoide in sede meta-diafisaria femorale omolaterale con assottigliamento della corticale e lieve rigonfiamento per-sottotrocanterico. L'epifisi appare risparmiata.
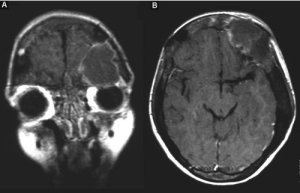 Fig. 3
Fig. 3 RM dopo m.d.c.:
A) migliore definizione dei rapporti anatomici della lesione pseudo-cistica fronto-orbitaria sinistra, complicata da fenomeni degenerativo-emorragici;
B) diffuso ispessimento e disomogenea ipointensità della teca.
